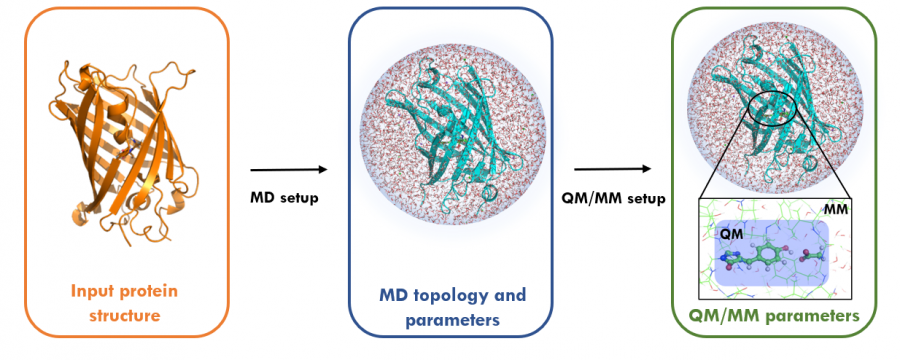
Quantum Mechanics/Molecular Mechanics approach
Unfortunately, the Quantum Mechanics approach does not scale up well. In particular, the CPU time increases typically as the 3rd power with the number of bases set functions in DFT-based methods. Fortunately, combining the Quantum Mechanics and Molecular Mechanics approaches can produce more accurate and efficient simulations.
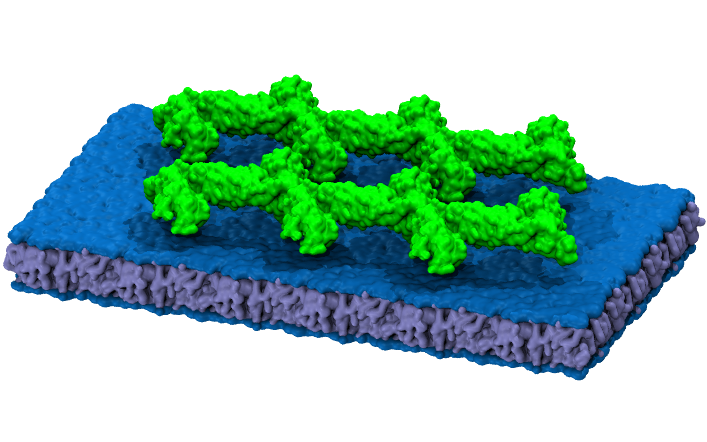
In QM/MM simulations, a small part of the system is treated quantum-mechanically. The remaining system is treated classically, thus allowing for studying chemical processes in solution and proteins or other complex interactions.
Applications of QM/MM include hybrid simulations of systems in systems where chemical reactions occur.
Visualisation and data analysis.
Data analysis for simulations can be carried out with a variety of software packages, for example:
Visual Molecular Dynamics (VMD) is a molecular modelling and visualisation computer program.
UCSF Chimera is an extensible program for interactive visualisation and analysis of molecular structures, including density maps, supramolecular assemblies, sequence alignments, docking results, trajectories, and conformational ensembles.
MDANSE (Molecular Dynamics Analysis for Neutron Scattering Experiments) is a python application designed for computing properties that can be directly compared with neutron scattering experiments such as the coherent and incoherent intermediate scattering functions and their Fourier transforms.