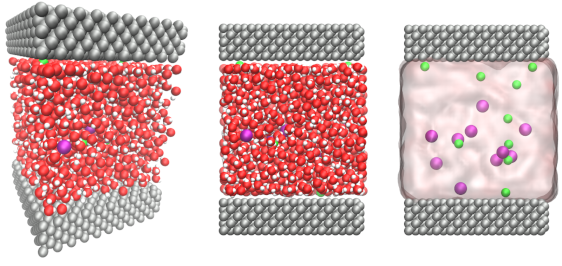

Large-scale Atomic Molecular Massively Parallel Simulator.
LAMMPS is a classical molecular dynamics simulation software that is useful for materials modelling. LAMMPS can complement neutron scattering experiments by helping visualise complex structures and interactions between atoms.
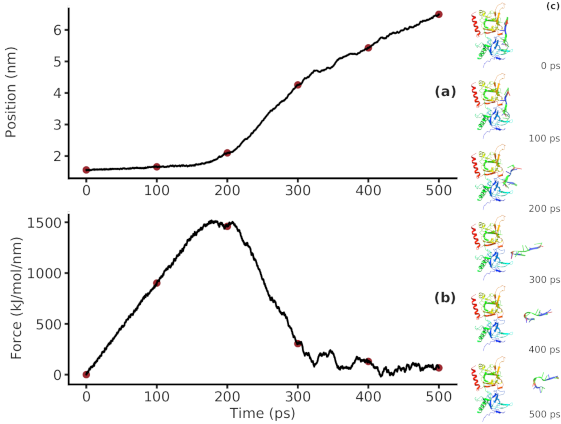
Groningen Machine for Chemical Simulations
GROMACS is a molecular dynamics package that simulates proteins, lipids, and nucleic acids. It can be used in conjunction with experiments to estimate binding energies between complex molecules and explore different thermodynamic configurations.
Contact us
